- Published on
- 16 min read
Designing gRNAs for base editing: hitting the right editing window
- Authors

- Name
- BioTech Bench
- @BiotechBench
Table of contents
- What you'll learn
- The editing window constraint: a different design mindset
- Counting positions manually
- CBE walkthrough: introducing a stop codon in TP53
- ABE walkthrough: correcting HFE C282Y
- Handling bystander edits
- PAM availability and what to do when you're stuck
- Common mistakes
- What's next
- Join the conversation
- Resources
This is Arc 1, Part 6 of the CRISPR from Bench to Analysis series.
You have a target base. You know CBE converts C→T and ABE converts A→G. You open a guide design tool, run your gene, and get 20 ranked guide candidates. Guide #1 scores 0.72. But when you check whether your target C is in the editing window, it's at position 2 — two positions before the window even starts.
Guide #12 scores 0.44 and puts your target C at position 6, alone in the window. Which guide do you use?
Guide #12. Every time.
This post teaches you why — and shows you how to find that guide reliably.
What you'll learn
- How to count protospacer positions and locate your target base in the editing window
- How the window constraint changes guide selection logic vs standard Cas9 design
- CBE walkthrough in BE-Designer: introducing a stop codon in TP53
- ABE walkthrough in BE-Designer: correcting HFE C282Y (hereditary hemochromatosis)
- How to handle bystander edits — and when to switch to a narrower-window editor variant
- How PAM availability affects base editing targeting options
The editing window constraint: a different design mindset
If you read Post 3 on base editing, you know the core mechanism: the deaminase domain of a base editor acts on single-stranded DNA exposed in the R-loop during Cas9 binding. Not all of that single-stranded DNA is accessible, though. The deaminase reaches only a defined stretch — roughly positions 4–8 from the PAM-distal end of the spacer, where position 1 is the first nucleotide of the spacer (the 5' end, furthest from the PAM) and position 20 is the nucleotide immediately before the PAM. This is the editing window. If you haven't read Post 3, that's the right place to start — this post builds directly on it.
The key design implication is a shift in logic. For standard Cas9 knockouts, on-target score is your primary filter: you want the guide that Cas9 binds most efficiently and that has the best specificity profile against off-targets. For base editing, window placement comes first. A high-scoring guide that places your target base outside positions 4–8 will not edit that base — full stop. The priority order for base editing guide selection is:
- Hard filters: GC 40–70%, no poly-T run (>4 T's in a row), no strong hairpin (same criteria as Post 5 — not repeated here)
- Window check: target base at positions 4–8 (CBE) or 4–7 (ABE7.10) / 4–8 (ABE8e). The 4–7 window applies to ABE7.10; ABE8e has a slightly wider 4–8 window. Check the spec sheet for whichever ABE variant you are using.
- Bystander check: no other editable bases (C for CBE, A for ABE) in the editing window
- On-target score: rank the surviving guides
Note the last step: score is used to rank guides that have already passed the window and bystander filters — not to select among all candidates. This is a fundamentally different use of the score.
Standard Cas9 guide design tools — CRISPOR, CRISPick (Broad Institute) — don't have dedicated base editing modes. They were built for knockout applications and output guide scores optimized for Cas9 cutting efficiency. CRISPick in particular only supports CRISPRko, CRISPRa, and CRISPRi modes; there is no base editing option. For base editing, you need a tool that explicitly shows you which bases fall in the editing window for every candidate guide. BE-Designer (CRISPR RGEN Tools) does exactly that. It's free, web-based, and purpose-built for CBE and ABE design. That's what we'll use for both examples in this post.
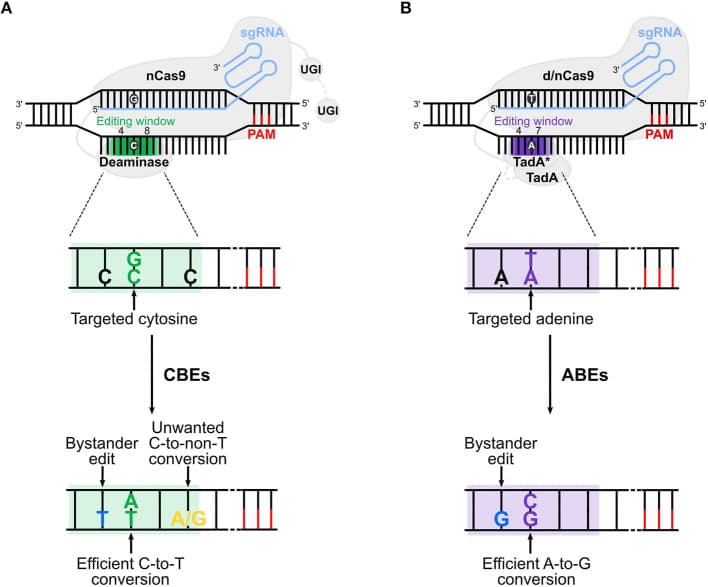
Figure 1. Cytosine base editors (CBE) and adenine base editors (ABE) act within the editing window — positions 4–8 from the PAM-distal end (green). A base outside the window will not be edited, regardless of how well the guide scores. Adapted from Antoniou P et al. (2021). Base and Prime Editing Technologies for Blood Disorders. Frontiers in Genome Editing, 3:618406. doi:10.3389/fgeed.2021.618406, under CC BY 4.0.
Counting positions manually
Numbering the protospacer is straightforward, but getting the direction right matters. Position 1 is the PAM-distal end — the 5' end of the spacer, the nucleotide furthest from the PAM. Position 20 is the PAM-proximal end — the last nucleotide before the NGG. Here is what that looks like with two cytosines in the protospacer:
5'- N N N C N C N N N N N N N N N N N N N N -3' NGG
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 PAM
CBE editing window: positions 4–8
C at position 4: ✓ in window — will be edited by CBE
C at position 6: ⚠ also in window — bystander risk
Both cytosines fall in the CBE editing window. If your target is the C at position 4, the C at position 6 is a bystander — it may also be converted to T, giving you a second, unintended mutation. This is the central bystander challenge in base editing, and it's addressed in more detail later in this post.
The most common counting error is starting from the wrong end — numbering from the PAM-proximal end instead of the PAM-distal end. If you make that mistake, you effectively place the window at positions 13–17, and every guide appears to miss. Always confirm: position 1 = 5' end of spacer = furthest from PAM.
One more thing to check when switching between editor types: the ABE7.10 window is one position narrower — positions 4–7. ABE8e, the higher-efficiency successor, uses a slightly wider 4–8 window matching CBE. Check your specific ABE variant's spec sheet before counting. The examples in this post use ABE7.10 as the reference. An adenine at position 8 is within the CBE window (if you were doing C-to-T editing nearby) but outside the ABE7.10 window. This is an easy mistake when you've been working in CBE mode and switch to ABE — recheck the window boundaries every time you change editor type.
CBE walkthrough: introducing a stop codon in TP53
A common CBE application in research is introducing a premature stop codon to generate defined loss-of-function alleles. The advantage over standard Cas9 knockout is predictability: instead of relying on the unpredictable indel spectrum from NHEJ, you get a defined C→T change that converts a glutamine (CAA or CAG) to a stop codon (TAA or TAG). TP53 exon 5 contains multiple glutamine codons and is a heavily studied region — it's a good practical target.
Input to BE-Designer:
- Go to BE-Designer
- Select: Nuclease = SpCas9, Base editor = CBE, PAM = NGG
- Paste the TP53 exon 5 genomic sequence (hg38) containing your target glutamine codon
- Organism: Human
- Run
To make this concrete: here is an example guide targeting a glutamine codon in TP53 exon 5. This is an illustrative example — retrieve your specific validated guide from BE-Designer.
Illustrative spacer (5'→3'): CCAGGGCAGCTACGGTTTCC
PAM: CGG
5'- C C A G G G C A G C T A C G G T T T C C -3' CGG
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 PAM
CBE editing window: positions 4–8
C at position 7: ✓ — this is the C in the CAG (glutamine) codon → TAG (stop)
C at position 1: ✗ — outside the window, not edited
C at position 20: ✗ — outside the window, not edited
Result: only one C in the editing window (position 7), no bystanders. This is exactly what you are looking for.
Reading the output — key columns:
| Column | What it means | What to look for |
|---|---|---|
| Guide sequence | 20-nt spacer | Check GC 40–70%, no poly-T |
| Editable bases | Cs in the editing window (positions 4–8) | Target C must appear here |
| Position | Position number of each editable base | Must be 4–8 for CBE |
| Off-target score | MIT specificity score | ≥70 for most applications |
| GC content | Spacer GC% | 40–70% |
Filtering workflow:
- Exclude guides with GC% outside 40–70%
- Exclude guides with poly-T warnings
- Filter to guides where the target C (from your glutamine CAA/CAG codon) appears in positions 4–8
- Among those, prefer guides with no other Cs in positions 4–8 (zero bystanders)
- If bystanders are unavoidable: note their positions and consider whether those bystander C→T changes are tolerable — are they synonymous? Do they hit an important residue?
- Sort remaining guides by off-target score; shortlist the top 3 for bench testing
For TP53 specifically, consider glutamine at codon 165 (CAG). A guide with an NGG PAM downstream can place the C of that CAG codon at one of positions 4–8 of the protospacer. BE-Designer will show the exact position for every candidate guide automatically — you don't need to calculate this by hand.
A note on opposite-strand targeting: if no guide on the coding strand places your target C in the window, check the template strand. A G on the coding strand is a C on the template strand. A CBE guide targeting the template strand can reach that base. BE-Designer searches both strands by default — confirm you're looking at both in the output. This step alone doubles your targeting options and resolves a surprising number of difficult cases.
ABE walkthrough: correcting HFE C282Y
The target: HFE C282Y (c.845G>A) is the most common pathogenic variant in hereditary hemochromatosis — the most common autosomal recessive disease in European populations. Homozygotes occur in approximately 1 in 200 people of Northern European descent and are at risk for iron overload that, if untreated, can progress to cirrhosis, diabetes, and cardiomyopathy.
C282Y denotes a cysteine-to-tyrosine substitution at residue 282 — the disease allele encodes tyrosine where the wild-type protein has cysteine (c.845G>A on the coding strand). At the DNA level, the disease allele carries an A at coding position 845; the wild-type sequence has G. ABE converts A→G, which directly restores the wild-type G — no donor template needed, no double-strand break required.
The guide:
Spacer (5'→3'): ACGTGCCAGGTGGAGCACCC
PAM: AGG
5'- A C G T G C C A G G T G G A G C A C C C -3' AGG
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 PAM
ABE editing window: positions 4–7
A at position 5: ✓ target base — HFE C282Y mutant A, corrected to G by ABE
No other A's at positions 4–7: zero bystander risk
The A at position 5 is the only adenine in the ABE editing window. No bystander adenines at positions 4, 6, or 7. This is an ideal base editing guide — a single, unambiguous edit.
This spacer is consistent with the HFE locus and the guide design used to demonstrate ABE in Gaudelli et al. 2017 (Nature 551:464). Retrieve and validate your final guide sequence in BE-Designer before ordering. It's a real-world example of why ABE's narrower window (4–7) can actually be an advantage: there are fewer positions that could harbor bystander adenines.
Using BE-Designer for this target:
- Go to BE-Designer
- Select: Nuclease = SpCas9, Base editor = ABE, PAM = NGG
- Paste the HFE genomic sequence around codon 282
- Run
BE-Designer will output all candidate guides with adenine positions highlighted. When it returns the guide above, you'll see position 5 flagged as the editable A — exactly what you want. The same filtering logic applies:
- Hard filters: GC 40–70%, no poly-T
- Target A at positions 4–7
- Zero bystander A's in the window
- Off-target score ≥70
- Rank survivors by on-target score
Window boundary reminder: ABE uses positions 4–7, not 4–8. An A at position 8 is outside the ABE window. Always recheck window boundaries when switching from CBE to ABE mode.
Handling bystander edits
A bystander edit is an unintended C→T (CBE) or A→G (ABE) change at a non-target position within the editing window. Any C at positions 4–8 is a CBE bystander candidate; any A at positions 4–7 is an ABE bystander candidate. This is the main practical limitation of base editing — and why guide selection is more constrained than for standard Cas9 knockouts.
Three strategies, in order of preference:
Strategy 1: Try alternative guides or the opposite strand
Before anything else, check whether a different guide — or targeting the complementary strand — places the target base alone in the window. If your target is G on the coding strand, it's C on the template strand; a CBE guide targeting the template strand can reach it. This step is zero-cost in BE-Designer (it returns both-strand results automatically) and solves the problem more often than expected. Exhaust this option before moving to strategies 2 or 3.
Strategy 2: Switch to a narrow-window editor variant
If no alternative guide gives a clean window, a narrower-window editor variant can restrict deaminase activity to fewer positions:
| Editor | Type | Approx. window | Notes |
|---|---|---|---|
| BE4max | CBE | Positions 4–8 | Standard CBE |
| YE1-BE4max | CBE | Positions 5–6 | Narrower, fewer bystanders |
| eA3A-BE3 | CBE | Position 5 (tight) | Very narrow; use when P5 is sole target |
| ABE7.10 | ABE | Positions 4–7 | Original ABE, most characterized |
| ABE8e | ABE | Positions 4–8 | High efficiency, slightly wider than original ABE |
| ABE8.20m | ABE | Narrower than ABE8e | Reduced bystander activity |
Window positions are approximate and sequence-context-dependent — always verify empirically. All major variants are available on Addgene.
Strategy 3: Accept bystanders and screen clones
If neither strategy 1 nor 2 resolves the problem, proceed with editing and sequence the full editing window in individual clones. Select clones where only the intended base was converted. This adds sequencing burden but is standard practice when bystander-free guides aren't available.
Whether a bystander is tolerable depends on context. A synonymous change in a non-essential region is usually acceptable. A bystander that creates a second missense mutation in a critical domain is not. Make this judgment explicitly before you run the experiment — not after you're looking at sequencing data.
PAM availability and what to do when you're stuck
Sometimes there is no SpCas9 guide that simultaneously places your target base in the editing window, avoids bystanders, and has an acceptable off-target profile. The constraint isn't the editor — it's PAM availability. SpCas9 requires NGG, and NGG sites are not uniformly distributed.
Option 1 — Expanded-PAM base editors: SpCas9-NG (recognizes NG PAM) and SpRY (recognizes NRN and NYN PAMs (near-PAMless)) have both been fused to CBE and ABE deaminases and are available as base editor constructs. They dramatically expand the set of targetable positions by relaxing the PAM requirement. The trade-off is real: lower on-target efficiency compared to standard SpCas9, and SpRY in particular has elevated off-target activity. Use these as a fallback when standard SpCas9 has no solution — not as a first choice.
Option 2 — Retarget within your region: For stop codon introduction, you're not locked to a single codon. Check all glutamine, arginine, and tryptophan codons in your target region — any of them could give you a tractable PAM and clean window. For precise mutation correction (like HFE C282Y), the target base is fixed, so you have less flexibility. In that case, expanded-PAM editors become more important.
Post 8 covers off-target prediction in detail, including SpRY-specific considerations and how to assess off-target risk when you have to use an expanded-PAM system.
Common mistakes
Counting positions from the PAM-proximal end. Position 1 is the nucleotide furthest from the PAM — the 5' end of the spacer. The editing window is positions 4–8 from that end. Counting from the wrong end places the window at positions 13–17 and no guide appears to work.
Forgetting to check both strands. If your target is G on the coding strand, it's C on the template strand — a CBE guide targeting the template strand can hit it. Only checking one strand halves your targeting options. BE-Designer searches both strands by default; confirm both are visible in the output.
Using on-target score as the primary filter. For base editing, window placement is the primary filter. A high-scoring guide that misses the window is worthless. Score ranks the window-compatible survivors — apply it last.
Assuming the same guide works for CBE and ABE. Window positions differ: 4–8 for CBE, 4–7 for ABE. A guide that places your target base at position 8 works for CBE but misses the ABE window. Recheck every guide when switching editor types.
Overlooking bystander bases on the non-template strand. Some CBE variants — particularly earlier-generation editors like BE3 — can deaminate cytosines in RNA transcripts or on the non-displaced strand under certain conditions. For precision applications, especially therapeutic use, validate the full editing window, not just the canonical spacer positions 4–8.
What's next
Post 7 — pegRNA design for prime editing. Prime editing has no editing window constraint; the RT template encodes the exact change you want. But pegRNA design introduces new complexity: primer binding site length, RT template design, the spacer-PBS junction, and nick guide selection. The design logic is structurally different from everything in Posts 5 and 6.
→ Next: Designing pegRNAs for prime editing: tools and principles
← Previous: What makes a good gRNA: Cas9 and Cas12a guide design principles
Want the complete base editing guide selection worksheet, bystander analysis protocol, and editor variant decision flowchart? They're in the book CRISPR from Bench to Analysis.
Join the conversation
Are you designing for loss-of-function (stop codon introduction) or precise mutation correction? Those two goals drive different bystander tolerance levels and editor variant choices — drop a comment below with what you're working on.
Resources
| Resource | What it's for | Link |
|---|---|---|
| BE-Designer (RGEN) | CBE and ABE guide design with window and bystander analysis | rgenome.net/be-designer |
| Komor et al. 2016 | Original CBE paper | Nature 533:420–424 |
| Gaudelli et al. 2017 | Original ABE paper + HFE C282Y validation | Nature 551:464–471 |
| Anzalone et al. 2020 | Base and prime editing review | Nature Biotechnology 38:824–844 |
| Antoniou et al. 2021 | Base editing in blood disorders (Figure 1 source) | Frontiers in Genome Editing 3:618406 |
Keep reading
On-target assessment methods compared: T7E1, TIDE, and amplicon NGS
You’ve made your edit, but how much of it is actually there? This post compares the three most common ways to measure CRISPR on-target efficiency—from the "quick and dirty" T7E1 gel assay to the high-resolution precision of Amplicon NGS.
Choosing your CRISPR system and designing validated guides: an end-to-end walkthrough
You have a gene to edit. Should you use Cas9, Cas12a, or a Base Editor? This capstone post walks you through the entire workflow—from choosing the right molecular "scissors" for your target to designing and validating your final guides.
In silico off-target prediction: how the algorithms work and which tools to trust
Choosing a guide RNA that cuts your target is only half the battle. You also need one that doesn’t cut anywhere else. This post breaks down the MIT and CFD scoring systems, how off-target algorithms actually work, and which tools are best for predicting Cas9 and Cas12a off-targets.